
{kind=link}
Factors Which Lead to increased Risk of Anemia
Intestinal disorders: Having an intestinal disorder that affects the absorption of nutrients in small intestine such as Crohn’s disease and celiac disease. Surgical removal or surgery to the parts of small intestine where nutrients are absorbed can lead to nutrient deficiencies and anemia.
Menstruation: In general, women who have not experienced menopause have a greater risk of iron deficiency anemia than do men and postmenopausal women. That is because menstruation causes the loss of red blood cells.
Pregnancy: In pregnancy, there is an increased risk of iron deficiency anemia because iron stores have to serve increased blood volume as well as be a source of hemoglobin for growing baby.
Family history: If family has a history of an inherited anemia, such as sickle cell anemia, the person may be at increased risk of the condition.
Other factors: A history of certain infections, blood diseases and autoimmune disorders, alcoholism, exposure to toxic chemicals, and the use of some medications can affect red blood cell production and lead to anemia.
Pathophysiology of Different Types of Anemia
[I] Pathophysiology of Iron Deficiency
Anemia: Iron is distributed in active metabolic and storage pools. Total body iron is about
3.5 g in healthy men and 2.5 g in women; the difference relates to women’s smaller body size, lower androgen levels, and lack of stored iron because of iron loss due to menses and pregnancy. Iron deficiency anemia is the most common form of anemia and it develops
over time if the body does not have enough iron to manufacture RBCs. Without enough iron, the body uses up all the iron it has stored in the liver, bone marrow and other organs. Once the stored iron is depleted, the body is able to make very few RBCs. If erythropoietin is present without sufficient iron, there is insufficient fuel for RBC production. The red blood cells that the body is able to make are abnormal and do not have a normal hemoglobin carrying capacity, as do normal red blood cells.
| Heme and Non-heme iron |
| Liver |
| Meats |
| Fish |
| Poultry |
| Non-heme Iron |
| Iron fortified infant formulas legumes |
| Eggs |
| Dairy foods |
| Wheat |
| Blackstrap molasses |
| Dried apricots |
| Raisins |
| Mustard greens |
| Strawberries |
| Tomatoes |
| Brussel sprouts |
| Broccoli |
Iron deficiency-consequences:
- Impaired physcial growth,
- Compromised cognitive development,
- Impaired learning capacity,
- Reduced muscle function,
- Decreased physical activity and
lower work productivity, - Lowered immunity,
- Increased risk of infectious disease.
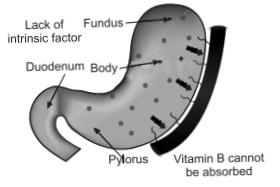
[II] Pathophysiology of Pernicious Anemia
In pernicious anemia vitamin B12 is unavailable due to a lack of intrinsic factor, a
substance responsible for intestinal absorption of the vitamin B12. In a healthy person, intrinsic factor is produced by the parietal cells of the stomach. Intrinsic factor forms a complex with dietary vitamin B12 in the stomach. This complex remains intact, preventing degradation of the vitamin by intestinal juices, until it reaches the ileum of the small intestine, where the vitamin is released and absorbed into the body. When intrinsic factor is prevented from binding with vitamin B12 or when the parietal cells are unable to produce intrinsic factor, the vitamin is not absorbed and pernicious anemia results. This is believed to stem from an autoimmune reaction in which the malfunctioning immune system produces antibodies against intrinsic factor and against the parietal cells. Without an adequate amount of vitamin B12, the body is unable to synthesize DNA properly. This in turn affects red blood cell production: the cells divide, but their nuclei remain immature, these cells are called as megaloblasts. Some megaloblasts mature to become large red blood cells are called macrocytes; they reach the circulation but function abnormally. A deficiency of white blood cells (leukopenia) and of platelets (thrombocytopenia) is also seen in the blood. Pernicious anemia occurs most often in persons older than 30 years of age, although a juvenile form of the disease does occur, usually in children younger than 3 years of age.
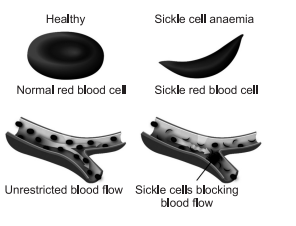
[III] Pathophysiology of Sickle Cell Anemia
The loss of red blood cell elasticity is central to the pathophysiology of sickle cell anemia. Normal red blood cells are quite elastic, which allows the cells to deform to pass through capillaries. In sickle cell anemia, low-oxygen tensions promotes red blood cell sickling and repeated episodes of sickling damage the cell membrane and decrease the cell’s elasticity. These cells fail to return to normal shape when normal oxygen tension is restored. As a consequence, these rigid blood cells are unable to deform as they pass through narrow capillaries, leading to vessel occlusion and ischaemia.
The actual anemia of the illness is caused by hemolysis, the destruction of the red cells inside the spleen, because of their mis-shape. Although the bone marrow attempts to compensate by creating new red cells, it does not match the rate of destruction. Healthy red blood cells typically live for 120 days, but sickle cells only survive 10–20 days.
[IV] Pathophysiology of Thalassemias
Normally, the majority of adult hemoglobin (HbA) is composed of four protein chains, two α and two β globin chains arranged into a heterotetramer. In thalassemia, patients have defects in either the α or β globin chain, causing production of abnormal red blood cells (In sickle-cell disease, the mutation is specific to β globin). The thalassemias are classified according to which chain of the hemoglobin molecule is affected. In α-thalassemias, production of the α-globin chain is affected, while in β thalassemia, production of the β-globin chain is affected.
| Structure: Smooth, flexible disk like structure that allows it to bend and flow through the blood vessels. | Structure : Sticky and stiff crescent shaped cell causing a lack of flow through the blood vessels. |
| Function: Can flow easily through the body and carry oxygen to all the organs. | Function: Can prohibit blood flow and cause pain to the person and damage to organs. |
| Hemoglobin: Normal iron rich protein that carries the oxygen throughout the body. | Hemoglobin: It causes the misshape and poor structure in the affected cell. |
| Recycle about every 120 days | Cells only live about 10-20 days |
The β-globin chains are encoded by a single gene on chromosome 11; α-globin chains are encoded by two closely linked genes on chromosome 16. Thus, in a normal person with two copies of each chromosome, two loci encode the β-chain, and four loci encode the α-chain. Deletion of one of the α loci has a high prevalence in people of African or Asian descent,
making them more likely to develop α-thalassemia. β-Thalassemias are not only common in Africans, but also in Greeks and Italians.
Related Feed:
Haematological Diseases | Anemia Disorders Conditions | Hematology Anemia Rash
Chronic Renal /Kidney Failure Symptoms | Diagnosis & Treatment Drugs
Chronic Renal Failure-Definition | Causes & Pathophysiology Of CRF
Acute Kidney Failure/Renal Diagnosis| Symptoms| Treatments and Drugs
Diagnosis & Treatment Chronic Obstructive Airways Diseases (COPD)